�Ķ�������141 ����ʱ�䣺2022/3/22 17:25:01
MAPKͨ·
MAPK��һ��˿/�հ��ᵰ��ø��Ŀ�Ѽ�����6���Ա���ֱ���ϸ������ڵ���ø(extracellular signal-regulated kinase, ERK)1/2��ERK3/4�� ERK5��ERK7/8��Jun����ĩ�˼�ø(Jun N-terminal kinase, JNK)1/2/3��p38 ��/β/γ(ERK6)/δ����ͬMAPK�������MAPK��ø(MAPKK)��MAPK��ø��ø(MAPKKK)��ϵ���γɱ��ص���ø������Ӧ(MAPKKK→MAPKK→MAPK)�����ε��ź�ͨ��������Ӧ��MAPK���������εĺ�ת¼���ӡ�ϸ���Ǽܵ��ף��γ�������MAPK�ź�ͨ·�������ϸ��������ĵ��ڡ�������MAPKͨ·�У�ERK1/2ͨ·�о���Ϊ���룬�방֢�Ĺ�ϵҲΪ���С�JNKͨ·��P38ͨ·�ڵ���ϸ������֢�������Ȼ�о�����Ҫ���ã�����������ͨ·���Դٽ���ϸ���ĵ������ٴ���Ӧ��As₂O₃
���ư�Ѫ������ͨ���JNKͨ·ʵ�֣�ŵ���������ɼ����Ϊ����ҩ����ͨ���P38ͨ·���ư�֢��
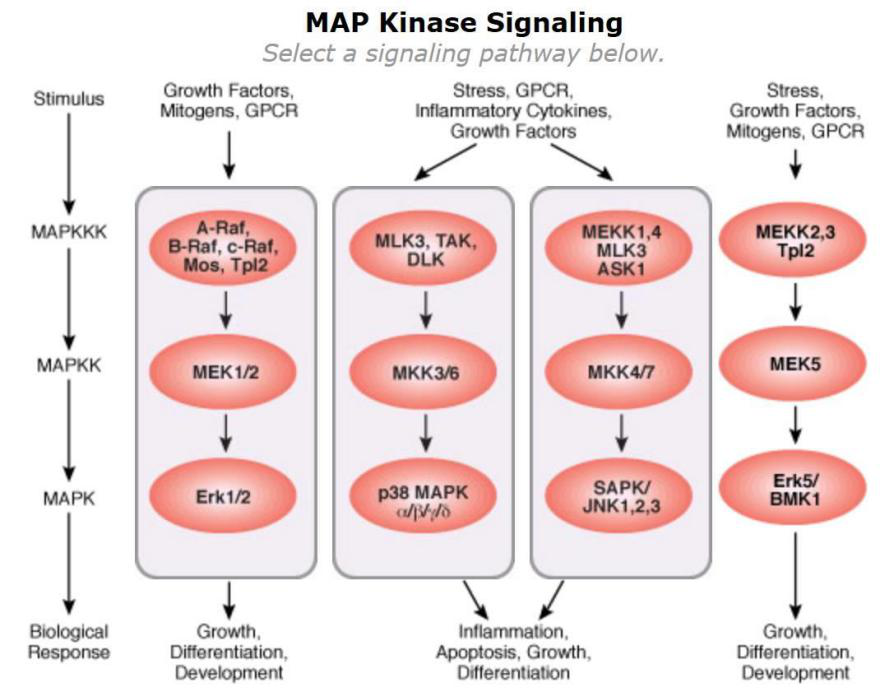
MAPKͨ·
ERK1/2�ź�ͨ·
ERK1/2�ź�ͨ·�ڵ���ϸ������ֳ���ֻ���������Ǩ�ƵȻ�з�����Ҫ�����á�������ź����������ӡ���˿����ԭ���������Ϻ��������Ұ��ἤø���ԣ�ͨ�������νӵ����źŴ��ݸ�СGTPøRas��Ras��GDP�����ת��ΪGTP����ͣ���������Raf�����RafʹMEK1/2�ϵ�˿����λ�����ữ������������ữERK1/2�ϵ��հ�����Ұ���л���������ERK1/2��һ�������ת¼���ӻ��߰�����������һϵ�еı仯������������У��κ�һ�ĵ��ױ��춼�п��ܵ��°�֢�IJ������о����֣�ERKͨ·����ͻ�侭�����ڰ�֢�г��֣���Լ�����ֵ�������������������ͼ���ͻ���RAS����Լ��8%������������BRAF���쳣���
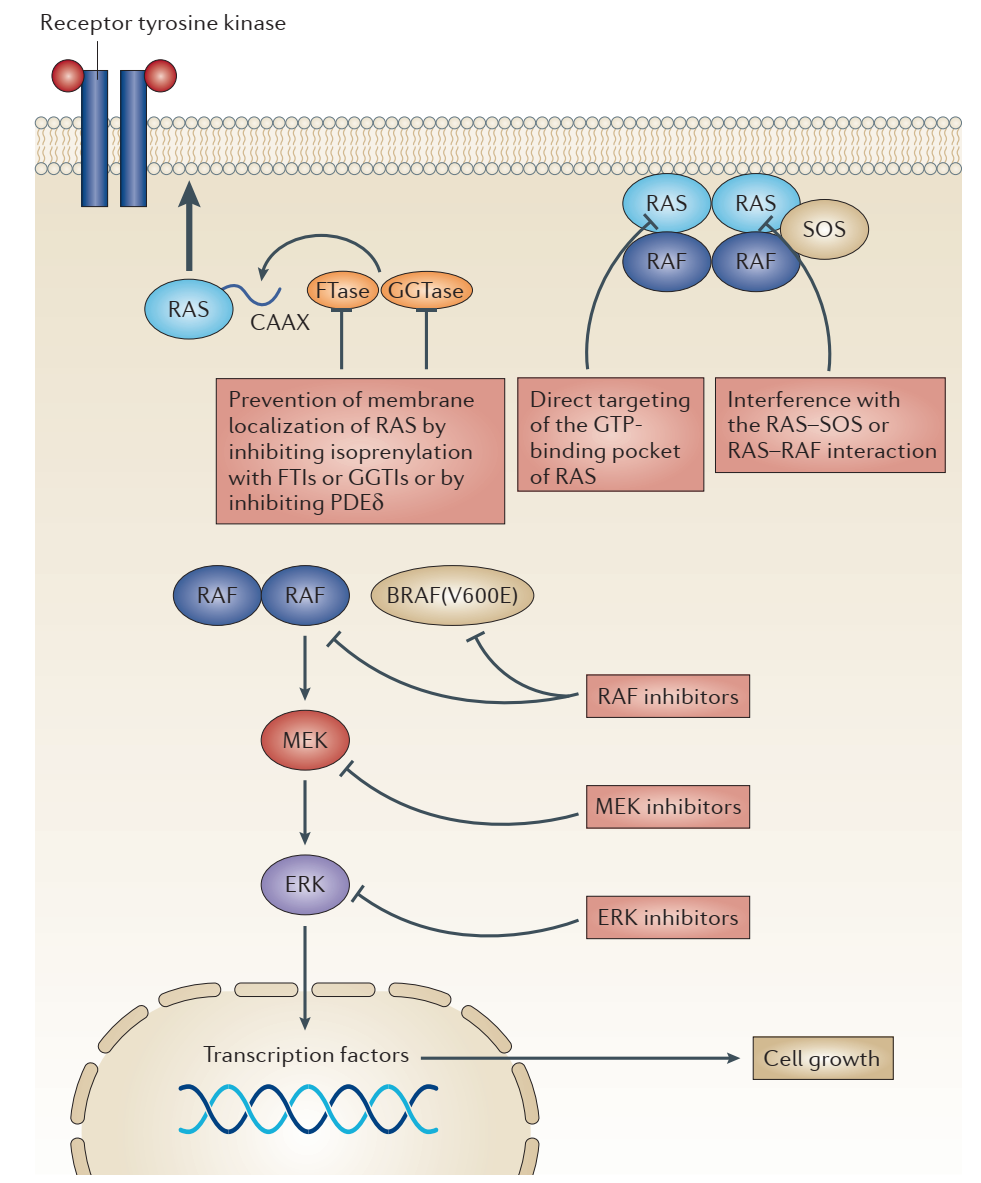
����ERK1/2�ź�ͨ·�Ŀ�������
����RAS��ҩ�↑������
��ȥ30�����������о������˲�ͬ����������RAS���嵰�ĸ�ͻ����(KRAS 20%, NRAS 8.0%, HRAS 3.3%)�������ü��嵰���ڰ�֢�����е�DZ�����á��о���Ҳ̽����RASͻ���°��Ļ������ƣ�������ͼ����RAS�ȶ���GTP��ϵĻ״̬���Ӷ�ʹ����ͨ·һֱ����������ϸ���Ĺ�����ֳ��Ȼ������RAS��GTP��ϵĸ���������������GTP��Ͽڴ���ҩ�↑��һֱ��һ�������ս��
RAS�����ϸ��Ĥ���伤��ı�Ҫ���̣��ɴ������һ����������ֹ����ϸ��Ĥ��ת�ơ������ת��ø�Ǵ�RAS�����ϸ��Ĥ�Ĺؼ�ø��Ȼ����Դ�ø���������Ƽ�(FTIs)Ч���������룬��ΪRAS��ϸ��Ĥ�Ľ��ͬ�����Ա���Ҷ����ת��ø����PDEδ�Ǵٽ�RAS��ȷ��λ��ϸ��Ĥ����Ҫ���ף����о�����������˵������Ƽ�����Ч����KRAS���°��źţ�Ȼ�����ֲ����ܷ������Ч�Ŀ���ҩ�ﻹ�д��ڹ۲졣
��������ֱ�Ӱ���RAS��ҩ�↑���ֱ������������𣬻��ڵ��ṹ��ƻ�����ķ������о��߿�������һϵ��RAS���ڼ���
�����ʽ�������SOS���Դ�RAS��GDP���״̬��GTP���״̬��ת��������Ƭ�λ�����ɸѡ�ķ������������о��ߵõ����ܹ���RAS��ϲ���������SOS����õĻ����Ȼ����������ЩС������RAS�ϵĽ�Ͽڴ��Ƚ�dz������ܷ�ͨ����һ�����Ż��õ��ٴ���Ч�Ļ�������Ȼ��һ�����⡣���⣬Ҳ���о���ͨ������ɸѡ���ֶζ����RAS-GTP��RAF��ϵ�С���ӽ�����ɸѡ����������һЩʵ����Ч�Ļ����Ȼ������Щ�������ѡ�����Լ���Ч����Ȼ���ɡ�
KRAS(G12C)ͻ���ǰ�֢�еij���ͻ�䣬���о��߱�����һ�����ڸ�ͻ�䵰�İ��װ��ᷴӦ�����Ƽ������ǵ��о�����������Ŀ�꣬�������ð��װ��ᷴӦ��С���ӻ������ɸѡ�õ���G12C����ϵ�С���ӣ����ڴ˻����ϵõ��ṹ�Ż���С���ӣ������ ��Ѱ��һ��������ͻ��RAS�ģ�����������������Ƽ���ͨ����G12C�����Ƽ����״̬�Ľṹ�о����֣��ҵ�����Щ���װ��ᷴӦ��С�����ǽ����RAS�ϵ��¿ڴ�����λ��ЧӦ����ϵĿ��آ������·�����Щ������Ľ�ϸı���RAS�ĺ�������ƫ�ã�ʹGDP�������GTP��ϣ��Ӷ�ʹRAS����ʧ��״̬������RAS���źš�Ŀ�����ڸò��Կ�����ҩ�����Lumakras (sotorasib, AMG510)��Adagrasib (MRTX849)�Ⱦ���ȡ�����õ��ٴ�Ч����
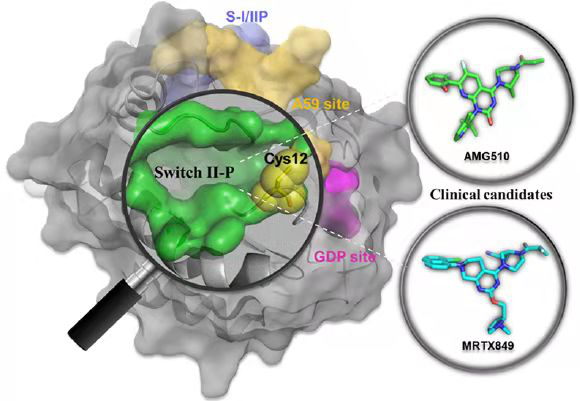
KRAS(G12C)ͻ�䵰���ϰ��װ��ᷴӦ��С���ӽ�Ͽڴ�
RAF�ڰ�֢�е�����
RAF��ø�������3�����ͣ��ֱ���ARAF��BRAF��CRAF�����Ǿ��и߶ȵ�ͬԴ�Ժ����ƵĽṹ��øC�˰����м�ø�ṹ��ȱ��RAS-GTPʱ��N�˻���C�˽��ʹ��ø���ڷǻ��Թ���
RAS�������RAS-GTP��RAF��RAS������ϡ�RAF�ļ�����һ�����ӵĹ��̣�����RAF����ļ��ϸ��Ĥ��RAS-RAF��������γɣ�RAF���������Ժ����Ե���λ������ữ��ȥ���ữ��RAF��ͬ���ۻ�������ۻ�������ȷ�е�˳�����ϸ��ϸ��Ŀ�Բ����������RAF���Ƽ������û��ƽ�ʾ�˶��ۻ���RAF���Ӵ������Լ�RAF���Ƽ��������õĹؼ����衣
CRAF���� һ������Ϊ����DZ���°����õ����ͣ�Χ�������о���ҩ�չ����Ҳ����չ���ġ�Ȼ�����֢�е�CRAFͻ��ʮ��ϡ�٣����ڿ�����CRAF���Ƽ���Ҫ��������ΪRASЧӦ���Ĺ��ܡ�
2002�꣬�о��������������з����˴�����BRAFͻ�䣬�о��Ľ���Ҳ��CRAFת����BRAF�������CRAF��BRAF���и��ߵĻ������ԡ���ˣ���һͻ������ʹBRAF�߶��쳣�����CRAF����Ҫ�����ͻ����ܴﵽͬ���Ļ��ԡ�
�Ӱ��ᵽ�Ȱ����ͻ�䣨V600E���dz�����BRAFͻ�䣬�������°�BRAFͻ����ռ����90%���ϡ��������°�BRAFͻ��ͨ���ۼ���BRAF�ļ���Ƭ�λ��߸����ʰ�����Ļ��С�
�����CRAF��BRAF��ARAF�ļ�ø���Խϵͣ�ARAFͻ���Ѿ���һС���ַΰ��Լ�����Langerhansϸ����֯ϸ������֢���ߵ���Ʒ�ּ���
��������е�RAF���Ƽ�——sorafenib����CRAF�㷺��BRAF���Ƽ���һ��������ٴ�����������������Я��BRAF(V600E)ͻ���ת���Ժ�ɫ������sorafenib��δ���ֳ���Ч�����á�
MEK�ڰ�֢�е�����
MEK1��MEK2��������ص�˫�������Լ�ø���鵼�����������źŴӼ���RAF��ERK�Ĵ�������Ȼ���������к��ٷ���MEK�������Ĵ��ڣ�������MEK����ͨ·��RAS��RAF�������Σ�ΪRAS��RAFͻ�䵼�������������ṩ��һ���µIJ��ԣ������һ�������������İе㡣Ŀ���Ѿ���ѡ���ԡ���ATP�����Ե�MEK�乹���Ƽ������ٴ���չ�Ρ������� һ�������ٴ���С�������Ƽ�CI-1040����Ȼ �����ڿڷ����Ժ���Ч�����Ⲣδ�������У��Լ��� һ����FDA����MEK1/MEK2���Ƽ� trametinib�ȡ�
ERK���Ƽ�
ERK1��ERK2���и߶�һ�µİ��������У�85%�������ǵ��Ұ�����հ���л��ɱ�MEK���ữ����������ERK���Ե���һϵ�е�ϸ���������ϸ����ֳ������ȡ�
��RAF��MEK��ȣ�ERK1��ERK2ѡ�������Ƽ��Ŀ����������룬һ����ԭ�������ڵļ�����ΪERK������MEK�����ΰб꣬ERK���Ƽ�������߱���MEK���Ƽ����������Ч����
Ȼ���������о����������ǿ���ERK���Ƽ�����Ȥ���ȣ�����RAF��MEK���Ƽ����о�ʹ������ʶ��������ͬһͨ·��ͬ��ֵ����Ƽ����Բ������Ӷ���������ѧЧӦ����Σ�ERK�ź�ͨ·��ͬ��ֵ����Ƽ��ٽ��ĸ�������·������ʾ����Ҫ�IJ��졣���⣬RAF��MEK���Ƽ�����ҩ��ͨ�����漰��ERK�ź�ͨ·�Ļָ���Ҳ����ERK���Ƽ���DZ����Ŀ������ERK��̱�����������SCH772984��BVD-523�� GDC0994 �ȡ�
�ܵ���˵��MAPK�ź�ͨ·������ERK1/2ͨ·�방֢�������е���ϵ�����ͨ·�йؼ�������ƵĿ���С����Ҳ��ʾ�����DZ���������о��IJ������룬�����ڲ��õ�δ�����Ὺ���������������Ч����С�������Ƽ�ҩ����Ź��˰�֢�ķ������һ����
���������ܹ��ṩ700����MAPKͨ·��ص�С�������Ƽ�������MAPKͨ·����о���
��ز�Ʒ��
AMG-510
AMG-510 is a selective ��nd orally bioavailable KRAS G12C covalent inhibitor.
MRTX849
MRTX849 is a potent, selective ��nd covalent KRASG12C inhibitor with potential antineoplastic activity. It selectively modifies mutant cysteine 12 in GDP-bound KRASG12C, ��nd inhibits KRAS-dependent signaling.
Sorafenib
Sorafenib is a potent multikinase inhibitor (IC50s: 6/20/22 nM for Raf-1/VEGFR-4/B-Raf).
CI1040
CI-1040 (PD184352) is an ATP non-competitive MEK1/2 inhibitor (IC50: 17 nM).
Trametinib
Trametinib is an ATP-noncompetitive inhibitor of MEK 1/2 (IC50s: 0.7/0.9 nM). It shows low inhibition for more than 180 kinases, including B-Raf, c-Raf, ��nd MEK5.
SCH772984
SCH 772984 is a potent inhibitor of ERK1/ERK2 (IC50: 4/1 nM) ��nd has only weak inhibitory for other 300 tested kinases.
Ulixertinib
Ulixertinib (BVD-523, VRT752271) is an effective ��nd reversible ERK1/ERK2 inhibitor. The IC50 of Ulixertinib is less than 0.3 nM for ERK2.
GDC-0994
GDC-0994 (Ravoxertinib) is an effective ��nd orally available ERK1/2 inhibitor (IC50: 1.1/0.3 nM).
����� - Samatar A A , Poulikakos P I . Targeting RAS-ERK signalling in cancer: promises ��nd challenges.[J]. Nature Reviews Drug Discovery, 2014, 13(12):928-42.
- FDA Approves First KRAS Inhibitor: Sotorasib. Cancer Discov. 2021 Jun 22. doi: 10.1158/2159-8290.CD-NB2021-0362. Epub ahead of print. PMID: 34158284.
- Lanman B A, Allen J R, Allen J G, et al. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors[J]. Journal of Medicinal Chemistry, 2019.
ԭ�����ߣ��Ϻ���������Ƽ�����˾






 Customer Service
Customer Service
